
二又研究室 Futamata Lab.
埼玉大学 理学部 基礎化学科
埼玉大学大学院 理工学研究科 化学系専攻
14
3.表面増強ラマン散乱(SERS)と表面増強赤外吸収(SEIRA)
赤外・ラマン分光の1分子当たりの信号検出感度を上げるためには、原理的に考えて、①入射光強度を大きくするか、②装置側でも分光器の効率及び検出器の感度を上げる、あるいは、③何らかの方法で試料からの信号強度の増強を行うしかない。このうち、①入射光強度を大きくすることは、赤外吸収ではまだ改善の余地があるが、ラマン散乱ではすでに100-1000 mWのレーザが実用されており、それ以上の入射光増大は、問題の表面化学種以外の光学素子、溶液セル、バルク種の蛍光やラマン散乱光を強めてしまい、効果的ではない。また、②装置の性能改善は進んでおり、赤外領域ではMCT検出器、可視紫外―近赤外領域ではCCD等のマルチチャンネル検出器の特性(感度、読み出し速度など)が、分光器の収差補正と共に十分に改善されてきた。これらに対して、③試料の信号強度については、化学種によらず10⁸ -10 倍の信号増強が実現できれば、赤外・ラマン分光でともに単一分子感度検出が可能となる。振動分光法の感度の改善に関しては、45年ほど前[6]に発見された原子レベルから100 nm程度の表面粗さを持つ金・銀・銅などの金属表面に吸着した化学種のラマン散乱が10⁴ -10⁶ 倍増強する表面増強ラマン散乱(surface enhanced Raman scattering, SERS)という現象が知られている(図➋-3, [7-9])。SERSと同等な赤外吸収の高感度化の手法として表面増強赤外吸収(surface enhanced infrared absorption, SEIRA, 図➋-4)が知られている。Osawaら[10]の開発したSEIRA法は、金属島状膜に吸着した化学種の赤外吸収を10-100倍増強することが知られており、特に電極表面で全反射配置(attenuated total reflection, ATR)を用いて詳細な研究が進められてきた。


3-1. SERS
表面増強ラマン散乱(surface enhanced Raman scattering, SERS)とは、原子レベルの粗さから100 nm程度の粗さを持つ金属表面―特にアルカリ金属や、金・銀・銅などの貨幣金属―に吸着した化学種のラマン散乱が10⁴ -10⁶ 倍増大する表面選択的な現象である。SERSは、1970年代初頭にFleischmannらにより偶然発見され[6]、当初表面粗さによる吸着有効面積の増大によるものとして説明されていたが、その後Creightonら[11]やvan Duyneら[12]により増強現象であることが確かめられた。増強メカニズムとともに、分析手法への応用を目指して、化学のみならず物理、生物系の科学者により盛んに研究がすすめられた。1980年代に、一旦第1次フィーバーが覚めた後、1990年に入ってからNie[13]やKneippら[14]により金ナノ粒子・銀ナノ粒子を用いて単一分子感度ラマンが実現できるという報告が出て以来、増強メカニズムの解明とともに、より大きな増強度を再現性良く与える金属ナノ構造やナノ構造体配列形成を目指した第2次フィーバーが2010年頃にかけて起こり、現在も下火になりながら続いている。
増強メカニズムとしては、当初から①化学的増強と②電磁気学的増強が、理論計算および実験により提案され、確かめられている(図➋-5A)。

①化学的増強メカニズムは、金属表面と吸着種との光誘起電子移動に基づく共鳴効果によるもので、10² -10³ 倍の増強を与える。電子移動のためには、金属表面に原子レベルの粗さを必要とし、吸着第1層の化学種にのみ活性を持つとともに、化学種依存性や振動モード選択性を有する([7], 図➋-6)。
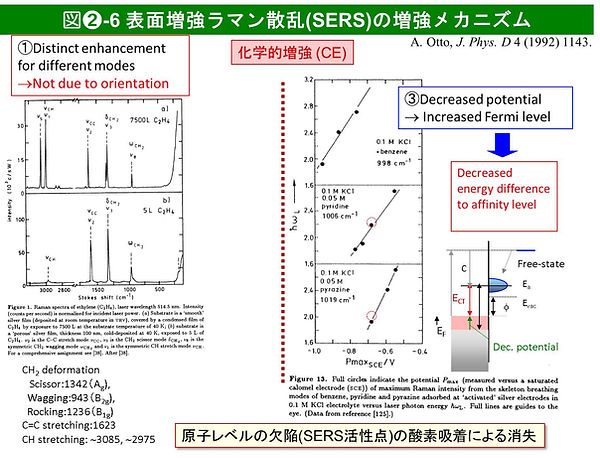
最近の研究によると、ナノ粒子を用いることで、励起電子の緩和時間を延ばし、電子移動が可能であり、化学的増強が平滑な表面のナノ粒子系でも起こり得ると考えられている。②電磁気学的増強は、当初、金属表面粗さに基づく局在表面プラズモン(localized surface plasmon, LSP)共鳴で形成される増強電場([7, 15])に加えて、全反射配置での伝搬性プラズモン(propagating surface plasmon, PSP)による電場増強が有効であることが、理論計算や実験により証明された。LSPの増強電場は、局所表面の曲率の逆数(r, 通常数nm-数10 nm)の距離で指数関数的に減衰する(表面選択性の原因)。PSPは、特にOtto配置では単結晶表面に適用できるもののLSPによるよりもやや小さな10² -10³ の増強を与える([16, 17], 図➋-7)。化学的増強とは異なり、電磁気学的増強メカニズムは、原理的に化学種によらず同等の増強度を与える。

Kneipp-Nie以後に主流となった金ナノ粒子や銀ナノ粒子(gold nanoparticle AuNP, silver nanoparticle AgNP, まとめて金属ナノ粒子MNP)を用いるSERSでは、MNPのLSP共鳴励起を利用する(図➋-8)。

孤立粒子のLSP励起では10⁴-10⁶ の粗い表面と同程度のラマン増強度(電場増強度は入射光の10² -10³ 倍)が得られる。これに対して、AuNP, AgNPを近接させ、粒子間ギャップが1 nm程度になると、個々のMNPのLSPがカップルして、ナノギャップに10⁴ -10⁵ の巨大な増強電場が形成される。このようなMNPの近接状態は、当初Siなどの基板上に、色素を吸着させたAgNP分散液を滴下・スピンコートし、偶然見つかるものを利用していた。我々は、カップルしたLSPの利用効率を上げるために、溶液中に孤立分散したMNP (AgNP, AuNP)とカチオン性色素、生体分子、水和金属イオン、アニオンなど種々の目的化学種との間の相互作用を制御し、近接状態を形成するflocculation-SERS法について検討を進めてきた([18], 図➋-8, -9A)。

近接状態の形成のために必要な金属ナノ粒子間相互作用と孤立分散安定性については次項(4. Flocculation-SERS法)で解説する。近接状態が形成されると、粒子内部に形成されるLSP双極子同士が、同位相で振動するために、粒子間ナノギャップに、粒子中心をつなぐ方向に平行な増強電場が形成されるため、粒子表面に吸着した化学種のラマン散乱が増強して検出される。このことは、(特に粒子サイズが<100 nmであれば)双極子-双極子相互作用により定性的に説明できる(>100 nmでは多極子LSPが励起される(図➋-8, -9B))。

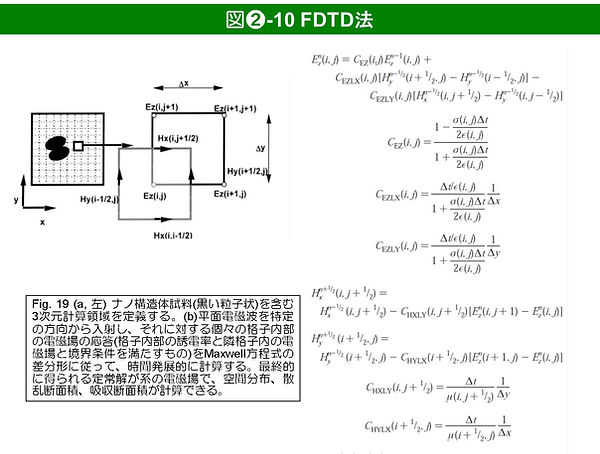
また、FDTD (finite-difference time-domain)法などの数値計算法により、定量的に増強電場の空間分布を得ることができる([19], 図➋-10)。
金属ナノ構造体間のカップリング(分極相互作用)の制御のために、電子ビームリソグラフィ(electron beam lithography, EBL)[22]が利用されている。近接した金属ナノ構造体配列の形成をめざしたものであるが、現状では電子ビーム径が10 nm程度であり、特別な工夫をしない限り、1-2 nmのナノギャップを持つナノ構造配列の形成は困難である(図➋-11)。
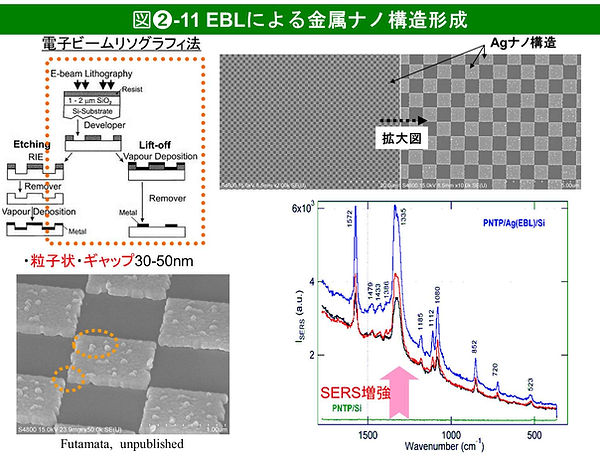
一般的なlift-off法に基づくEBL法では、①Si基板等に、一定膜厚のポリマーレジスト(PL)をスピンコートで形成、②電子ビームで、PL上に描画、③電子ビーム照射場所により結合が解列したPL部分を有機溶媒1で溶解除去、④その上に金属薄膜を真空蒸着で形成、⑤最後に、有機溶媒2で、残留しているPLをその上の金属薄膜ごと溶解除去するもので、結果的に最初に電子ビーム照射した領域に金属ナノ構造体配列が残る。このようにEBLでは、真空蒸着で金属薄膜を形成するために、基板表面にGe 下地膜を形成する等の処理をしない限り、粒子状成長した金属ナノ構造体が形成される。そのため、ナノ構造体配列によるLSPカップリングと、粒子状金属ナノ粒子自身のLSPによる効果が重なるので注意が必要である。
他方、化学還元法の精密な制御により、球状粒子、ロッド、立方体、星形など特徴的なナノ粒子を形成することが可能になり、表面粗さとある意味で等価な金属ナノ粒子(metal nanoparticle, MNP)及びその集合体(配列)を用いたSERS検出が盛んに研究されている(図➋-12, [20])

基本的に孤立状態でのラマン増強度は近接状態に比べてずっと小さいため、避雷針効果を併用する星型構造(nanostar)のような、複雑な構造体が形成されている。特にSERSを利用したバイオセンシング(in vitroでの細胞内の代謝等の静的・動的観察、生体内の腫瘍検出、ドラッグデリバリやサーモセラピー)、あるいは毒物検出など幅広く利用されている[21]。ただし、残念なことに、昨今の論文数のインフレ状況を反映して、多くのSERS研究論文はナノ構造体形成とそのSERS活性の確認で終わるか、または生体内のラマン信号を測定するのではなく、腫瘍等を認識するタンパクとともにラマンラベル用色素をつけた(tag)金ナノ粒子等を生体内に導入し、色素のラマンスペクトルの検出を行っている(図➋-13)。
これらのアプローチでは、非常に多くの類似した論文(例えば、ナノ粒子の材料・形状・サイズ・集合状態の違い、ラベル色素の違い、生体内ターゲット物質の違い、毒物の種類の違い、定量分析性、感度改善、デバイス化など)を生産することになる。これは、生体内のラマンスペクトルがきわめて複雑で、分子識別自体が容易ではないことから考えて、現実的な方法ではある。しかし、複雑な生体内現象の分子レベルでの理解にはなかなか近づけない。本来のラマン・赤外分光は、分子の指紋であり、直接対象分子のラマンスペクトル検出により、その識別や定量、状態分析を行うのが理想である。この点を改善しない限り、同じことを蛍光分光で行えるので、ラマン分光を利用する特別な理由はない。本質的な改善のためには、
①金属表面の分析対象(高)分子を含む化学種の吸着・脱離に関する表面化学の実験的解明が必須である。
②その上で、金属を含む吸着分子系の電子状態の解明:実験的には、UPS, 逆光電子分光などによるFermi準位と吸着種のHOMOおよびLUMOの解析、同時にDFT計算等による解析が必要である。
①表面化学(図➋-14):金属表面への溶液中化学種の吸着・脱離は、地道な解析によるしかない。例えば、化学還元で形成した金ナノ粒子表面にはクエン酸アニオンと塩化物イオンが吸着している。特にクエン酸イオンは、銀ナノ粒子系とは異なり、塩化物置換ができない。見かけ上、Au-citrate3-の相互作用がAu-Cl-、Ag-citrate3-の相互作用よりもずっと強い。静電的な相互作用であれば、pHを下げて、citrateにプロトン付加すればよさそうであるが、AuNP表面のcitrateは、pH2(バルク水溶液中のcitrateは完全にプロトン付加しcitric acidとなる)でも、-2価のイオンとして存在する。感度が大幅に改善されても、界面化学種の構造のわずかな変化や配向性を解析するには、重水素置換体や関連化合物とのスペクトル比較、pH依存性や溶液組成制御下でのスペクトル変化に加え、DFT計算、XPS測定やゼータ電位測定などの測定と解析が必要であり長時間を要するのが現状である。しかし、幅広い化学種に適用可能なSERSを(定量)状態分析法として確立するためには、こうした地道な努力が必須である。
②金属を含む吸着分子系の電子状態の解明:実験的には、UPS, 逆光電子分光などによるFermi準位と吸着種のHOMOおよびLUMOの解析により、光照射下でプラズモン励起に引き続いて、金属側からの電子(ホール)移動による吸着種の還元(酸化)が、エネルギー的に起こり得るかどうかを明らかにすることが必要である。同時にDFT計算等による金属と吸着種を統合した電子状態の解析、実験結果と対応づけた理解が必要である(図➋-15)。これらは化学的ぞきょうメカニズムに関係するとともに、最近注目されているプラズモンが関与する光触媒反応が起きるかどうか理解するためにも重要である。原理的・手法的には明確であるが、現時点では、いずれも十分な実験データ理論解析結果は限定的であり、発展途上にある。

3.2 SEIRA
金属の種類によらず、金・銀・銅以外にも、白金、パラジウムなど幅広い金属島状膜や金属ナノ粒子が、増強基板として用いられる[10]。当初、増強メカニズムは、真空蒸着で形成された島状膜では、金属ナノ粒子が扁平であり、基板に平行な方向に粒子間が近接していることに基づいて説明された。このとき、回転楕円体のLSPカップリングにより、共鳴波長が中赤外領域にシフトすることで、入射赤外光を増強し、吸着種の赤外吸収が増大する。確かに、局所的な電場増強は赤外増強吸収を生じる。最近のナノロッドの軸比を極端に大きくする方法で、赤外領域までLSP共鳴波長を伸ばし、Fano共鳴を用いてSEIRA効果を実証したとの報告もある[23]。最近、特異的なナノ構造体の近接配列などのメタマテリアル構造により赤外光領域で局所電場を増強できることが理論計算により示され、共鳴波長範囲は数100 cm⁻¹ 以下とやや狭いものの実験的に確かめられた[24]。これらは、基板表面に垂直な方向からの赤外光入射で有効に働く。実際に、電極表面系でよく利用されている全反射SEIRA配置では、基板に垂直な方向のエバネッセント波が形成されるために、こうした説明にはやや無理がある。この場合、誘電分散により、赤外光領域では、どの金属の誘電率も、実部(<0)・虚部ともに非常に大きな値を持つ。単純に、この誘電率の効果と、ナノ粒子間ギャップ付近の避雷針効果で、実測された10-100倍の赤外吸収の増強は得られる可能
Au連続膜とその上に>200 nmのやや大きめの粒子状Auが配置したギャップモードが、全反射配置で有効に働くものと考えられる。今後のメカニズム解明が待たれる。
参考文献
(6) M. Fleischmann, P. J. Hendra, A. J. McQuillan, Chemical Physics Letters 1974, 26, 163-166.
(7) A Otto, I Mrozek, H Grabhorn and W Akemann, J. Phys.: Condens. Matter 1992, 4 1143-1219.
(8) K. Kneipp, M. Moskovitz, H. Kneipp, “Surface Enhanced Raman Scattering”, Top. Appl. Phys. 103, 2006.
(9) Eric C. Le Ru and Pablo G. Etchegoin, “Principles of Surface-Enhanced Raman Spectroscopy”, Elsevier 2009.
(10) a) M. Osawa, Bull. Chem. Soc. Jpn. 1997, 70, 2861-2880. b) M. Osawa, Top. Appl. Phys. 2001, 81, 163-187.
(11) M.G. Albrecht, J. A. Creighton, J. Am. Chem. Soc. 1977, 99, 5215–7. (12) D. L. Jeanmaire, R. P. van Duyne, Journal of Electroanalytical Chemistry, 1977, 84, 1–20.
(13) S. Nie, S. R. Emory, Science, 1997, 275, 1102–6.
(14) K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari, M. S. Feld, Phys. Rev. Lett. 1997, 78, 1667.
(15) M. Kerker, “Selected Papers on Surface-Enhanced Raman Scattering”, SPIE 1990.
(16) K. Kurosawa, R. M. Pierce, S. Ushioda, Phys. Rev. B 1986, 33,789-798.
(17) M. Futamata, E. Keim, A. Bruckbauer, A. Otto, Appl. Surf. Sci. 1996, 100-101, 60-63.
(18) a) M. Futamata, Y. Yu, T. Yajima, J. Phys. Chem. C 2011, 115, 5271–5279. b) T. Yajima, Y. Yu, M. Futamata, Phys. Chem. Chem. Phys., 2011, 13, 12454–12462. c) T. Yajima, Y. Yu, M. Futamata, J. Raman Spectrosc. 2013, 44, 406–411, d) 半田紗織・Yingying YU・谷島徹・二又政之, 表面科学, 2013, 34, 449-454.
(19) M. Futamata, Y. Maruyama, M. Ishikawa, J. Phys. Chem. B 2003, 107, 7607-7617.
(20) N. Li, P. Zhao, D. Astruc, Angew. Chem. Int. Ed. 2014, 53, 1756 – 1789.
(21) K. Saha, S. S. Agasti, C. Kim, X. Li, V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779.
(22) R. F. Peters, L. Gutierrez-Rivera1, S. K. Dew, M. Stepanova, Journal of Visualized Experiments, 2015, 97, e52712.
(23) C. Huck, J. Vogt, M. Sendner, D. Hengstler, F. Neubrech, A. Pucci, ACS Photonics 2015, 2, 1489-1497.
(24) a) A. Ishikawa, S. Hara, T. Tanaka, Y. Hayashi, K. Tsuruta, Scientific Reports 2017, 7, 3205. b) I. M. Pryce, Y. A. Kelaita, K.Aydin, H. A. Atwater, ACS Nano, 2011, 5, 8167–8174.