
二又研究室 Futamata Lab.
埼玉大学 理学部 基礎化学科
埼玉大学大学院 理工学研究科 化学系専攻
4.金属ナノ粒子を利用したflocculation-SERS
粗さを持つ金属表面の代わりに、金属ナノ粒子を用いて表面増強ラマン散乱を得ることができる。金属ナノ粒子に光を照射すると、Mie散乱理論で与えられるように、電磁気学的な境界条件を満たす電磁場(固有モード)が、ナノ粒子に形成される[25]。これは、直感的には次のように説明できる。可視光の波長(400-700 nm)に比べて十分小さなナノ粒子(数10nm径)を用い、粒子固有の共鳴振動数(局在表面プラズモン, localized surface plasmon, LSP, Eq. 4-1)の光を照射すると、ナノ粒子内部の電子は場所によらず同じ位相で振動する。ある瞬間には粒子の片側に電子(負電荷)が偏在する状態(同時に粒子の反対側には正電荷が偏在)となる。この時、ナノ粒子に光双極子―光の振動数で周期的に変化するので、粒子内部には、振動する双極子―が形成される。振動双極子は、粒子近傍に局在する伝搬しないエバネッセント波(近接場光)と、遠方まで伝搬する電場(伝搬光)を生成する。これらの電場の時間・空間的な広がりや大きさは、古典電磁気学により明瞭に記述される(Eq. 4-2)。とくに、近接場光は入射光に比べてずっと大きな電場強度(AuNP, AgNPでは約10² 倍)を与える。ラマン散乱では、入射光の増強による効果とともに、ラマン散乱光自身も金属ナノ粒子により増強されるため、金属ナノ粒子表面に存在する吸着種のラマン散乱は、入射電場増強´散乱電場増強=10⁴ -10⁵ 倍も増強される。これは表面粗さを有する金や銀表面と同程度の増強である。この増強電場の大きさは、LSP共鳴波長で最大となる。LSP共鳴波長は、金属ナノ粒子を分散した水溶液を可視紫外分光計で測定することでextinction spectra (extinction = absorption + scattering, 粒子サイズが分子よりずっと大きくなると、吸収(absorption)と散乱(scattering)により入射光が減光(extinction)する。実際に、LSP共鳴波長では、吸収と散乱が起きる。この場合の吸収は、集団的な電子励起状態への遷移、散乱は、励起され形成された双極子からのいろいろな方向への弾性的散乱)として得られる。金属の誘電率、粒子サイズ、形状のほか媒質の誘電率により決まる。LSP共鳴波長は、<100 nmの大きさの球状銀ナノ粒子では400 nm、球状金ナノ粒子では525 nmであり、粒子サイズの増大に伴い、わずかに長波長シフトする。また、>100 nmでは双極子LSPのほかに、異なる共鳴波長を持った多極子LSPが励起され、電場の空間分布がそれに応じて変化する。回転楕円体など異方性を持った粒子では、粒子サイズが小さいうち(<100 nm)は、軸比とともに大きく長波長シフトする。サイズが大きい(>数100 nm)ロッド状粒子では、軸比ではなくサイズで共鳴波長が決まる。
先に挙げたNie, Kneippらは、単一分子検出のために必要なラマン増強を得るために、AuNP, AgNPの凝集体(近接体)が有効であることを示した。これは、ナノ粒子が凝集することで、個々のナノ粒子のLSPが結合(coupling, 直観的には同位相で振動する個々の粒子のLSPが干渉効果で、より大きな振幅を持つと解釈できる)し、粒子間ナノギャップで、10⁴ -10⁵ 倍の電場増強(10⁸ -10¹⁰ のラマン増強)が得られることを利用する。金属ナノ粒子の近接状態―数えられる程度の粒子がnmギャップを持って近接した状態-では、効率的なLSPカップリングが起きる。これに対し凝集体ではナノ粒子の一部が凝結・融合し、部分的に一体化してしまうため、原理的に増強度の制御や定量分析、吸着状態の解析などが困難である。次節ではどのようにして、金属ナノ粒子を近接させ、一定時間(少なくとも測定の間)安定に保つことができるかを示す。
4.1金属ナノ粒子間相互作用:金属ナノ粒子のflocculation (近接), aggregation (凝集)
金属ナノ粒子の近接状態を形成し、安定に保つためには、金属ナノ粒子の表面間力を制御する必要がある。SERS研究で広く用いられる金ナノ粒子(AuNP)や銀ナノ粒子(AgNP)は、多くの場合クエン酸化学還元法で形成される[26, 27]。このとき、金属ナノ粒子表面にはクエン酸イオンが残留吸着しており、負の表面電荷を持つ。そのためこれらの金属ナノ粒子(MNP)は相互に静電反発し、水溶液中で数ヶ月間にわたり安定に孤立分散する。こうした帯電ナノ粒子間には、Eq. 4-3式で示されるように、個々の粒子を構成する金属原子と別の粒子の金属原子間で働くvan der Waals引力の総和(原子間や分子間の場合と異なり、r⁻⁶ ではなく、r⁻¹ やr⁻² に比例する長距離力である, VvdW)とともに、ナノ粒子の表面電荷あるいは溶液中の対イオンが作る電気二重層間に働く斥力が働く(Vrepul, 図➋-16 [28])。

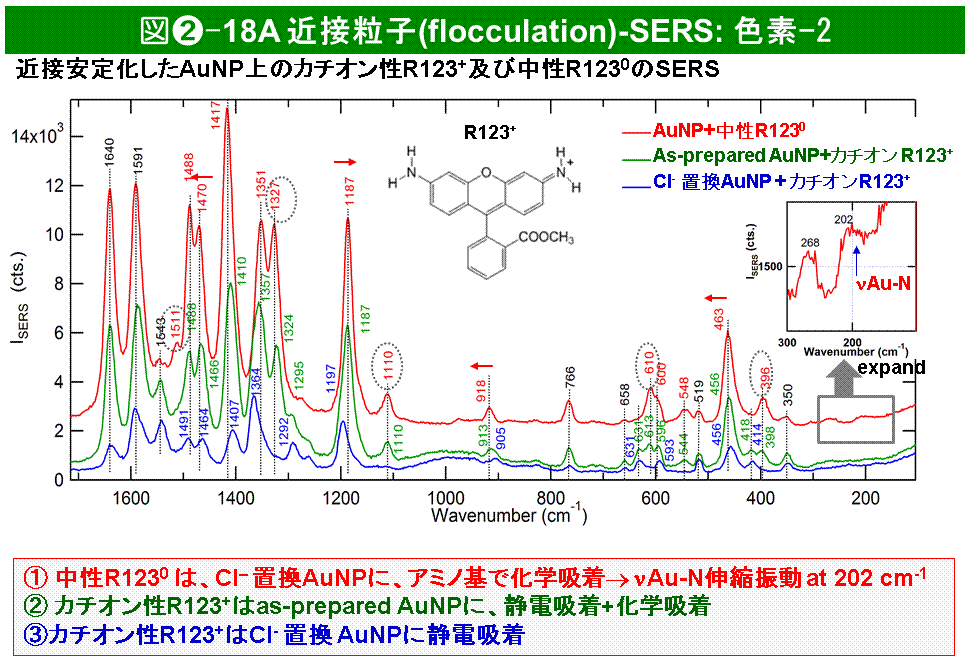
このうち、van der Waalsポテンシャル(VvdW)は、粒子の材質、大きさ、形状等で決まるものでHamaker定数を使って簡略に表される。静電反発ポテンシャル(Vrepul)は、イオンの周りの静電ポテンシャルと同様に、粒子表面の電荷密度、粒子間距離、媒質の誘電率、溶液のイオン強度等で決まる。特に、イオン強度の増大とともに、表面ポテンシャルの低下及び電気二重層厚さの現象のために、粒子間の静電斥力ポテンシャルVrepulはより短距離で低下する。そのため、全粒子間相互作用Vtotalにおいて、エネルギー障壁EBが低下する。すなわち、イオン強度が増大すると、粒子は熱エネルギーkBTによりEBを超える確率が増大し、近接しやすくなる。一旦近接(粒子間距離d<5 nm)すると、van der Waalsポテンシャルが支配し、熱エネルギーkBTで逆向きのバリアEB’を超えることは不可能である。近接した金属ナノ粒子は、そのまま静置すると、条件により異なるが数日のうちに、原子位置の交換を通して、凝結・融合し、デンドライト結晶などに変化する。塩濃度をccfよりずっと高くすると、瞬間的に凝集状態が形成される((図➋-17a, -17b, 写真)。この場合、吸着種も状態が変化したり、部分的に脱離してしまうと考えられる(凝集状態で得られたスペクトルと孤立粒子表面での吸着状態の同等性が保証されない)。しかし、濃度をccf近傍で適切に制御すると、凝集しないで、長時間にわたり限られた数のナノ粒子が近接した状態が保持される((図➋-17c, 写真)。MNPの孤立状態、近接状態、凝集状態は、extinction spectra測定により、識別できる(図➋-17d)。孤立状態では、MNPのLSPによるピーク(AgNPでは400 nm, AuNPでは525 nm付近)に1本のピークが観測される。一定時間保持後、LSPピークの吸収強度減少とともに、長波長側にショルダーまたはブロードなピークがあらわれはじめる濃度を近接臨界濃度(ccf, critical concentration for flocculation)と定義する。近接状態では、孤立粒子のLSPピークよりも長波長側にややブロードなピークが観測される。これは近接MNPにおいてカップリングしたLSPの励起によるものである。この近接粒子のピーク波長は、粒子間ナノギャップサイズが小さいほど長波長にシフトし、また1つの近接粒子群に存在する粒子数の増大とともに長波長シフトする[39]。凝集状態では、集合状態の中の粒子数の増大とともに、拡散速度の低下のために粒子は溶液下方に保持され、やがて沈殿するので、extinction spectraは、当初、孤立粒子・近接粒子のピークが弱くなり、ブロードなバックグラウンドを与えたのち、最後には溶液層から粒子が消失するため、無色透明となり、Flocculation-SERS法では、系によって決まる塩濃度や分析対象化学種のccfをまず実験的に決定し、これらの濃度をccf近傍に保つことで、2-数個の近接した金属ナノ粒子(MNP)を形成し、その粒子間ナノギャップに吸着した化学種のラマン散乱を高感度で検出することができる。例えば、ローダミンなどの色素分子の一分子感度検出も可能である([3], 図➋-18)。

他の分子系でも、DNA塩基([4], 図➋-19)、各種チオール(図➋-20)、水和金属イオン([5], 次項)など。

このような高感度ラマン分光測定で得られるラマンスペクトルの1本1本のピークは、すでに�述べた分子の1個1個の内部振動に対応している。それは分子内の結合の伸縮や、結合角の変角振動のエネルギーを示すので、吸着状態のラマンスペクトルを解析すると、界面での分子の構造変化や、表面に対する配向性、配位結合や静電的相互作用などの表面との結合相互作用のほか、他の分析法や理論計算と複合することで電子的な相互作用について情報を得ることができる。
Eq.4-3 ナノ粒子間相互作用=van der Waalsと静電反発式
金属ナノ粒子(半径r=r1, r2)間の相互作用(Vtotal = Vvdw + Velec)[文献]は、van der Waalsポテンシャル(Vvdw)とクーロンポテンシャル(Velec)の和として、次式で表わされる。
AH: ハマーカー定数(Au, Agで2×10 ⁻¹⁹ J)
x: ギャップサイズ(=粒子表面間距離)
D=r1+r2+x (=粒子間距離, nm)
k: Debye-Hückel定数
e=e0et, y0:MNP表面電位(V).

次に、当研究室で進めているflocculation-SERS法を適用した例をいくつか取り上げ、やや詳しく説明する。
4-2. 色素分子の吸着状態
4-3.水和金属イオンの捕捉と検出
最初の例は、水和金属イオンを銀ナノ粒子のflocculationを用い�て捕捉し、水和状態について新しい情報が得られるというものである[35]。一般に、溶液中の金属カチオンは、対アニオンとcontact ion pair (CIP; 裸のカチオンと裸のアニオンが形成する接触イオン対), solvent-shared ion pair (SIP; カチオンとアニオンの間に1層の溶媒分子を共有するイオン対), solvent separated ion pair (2SIP;溶媒和カチオンと溶媒和アニオンが、それぞれの溶媒和分子を持って形成するイオン対) の3種類のイオン対を形成することが、誘電緩和[29a]、X線散乱(XS, XANE, EXAFS[30])、振動分光[31]、MD計算[32]などに基づいて報告されている(図➋-21A~➋-21C, [29b])。


例えば、濃厚金属塩水溶液の赤外吸収スペクトル測定にもとづいて、金属イオンは+1価、+2価などあるいは、第1水和圏、第2水和圏などのクラスごとに、金属の種類によらずほぼ一定のO-H (O-D)伸縮振動が測定される一方、アニオンに溶媒和した水分子では、電荷密度(静電パラメータz=z/r, z:価数、r:イオン半径で表される)とともに大きくO-H伸縮振動数が異なることが報告されている[33]。定性的・直感的には、構造形成アニオンでは、第1層だけでなく第2層水和圏を形成し、それらの間の分子間水素結合が、バルク水に比べてより強く、バルク水のO-H伸縮振動より低波数ピークを与え、構造破壊アニオンでは、第1層水和層のみ形成し、水分子間水素結合はバルク水より弱まり、より高波数のO-H伸縮振動を与えると説明される。ただし、より厳密には水分子のO-H伸縮振動は、分子内カップリング(2本のO-H伸縮振動のカップリング及びO-H伸縮振動とH-O-H変角振動の倍音の非調和性によるFermi共鳴)、異なる水分子間のO-H伸縮振動のカップリング(クラスタ的な構造体を作るときに特に大きい)のほか、分子間水素結合の強さにより決まる複雑なものである。それらに加えて、数M以上の濃厚な溶液でない限り、水和水よりも大量に存在するバルク水分子の妨害が重なる。そのため、分子内カップリングを抑制するための部分的に重水素化した水分子H-O-Dを用いる方法や、通常ブロードでいくつかのピークの重なりとして得られるスペクトルの多変量解析などが用いられている。それでも、金属イオンとの水和水の構造や相互作用の理解は十分進んでいない。
-1

一方で、SERS発見以来45年に及ぶ研究の歴史の中で、様々な化学種の高感度ラマン検出が行われてきたが、水分子を検出した例は、電気化学的酸化還元処理で粗くした金、銀、白金電極表面での測定結果[34]以外、ほとんど報告例はない。これは、粗くした金属表面でのLSPによるSERS増強度が10⁴ -10⁵ と比較的小さいことと共に、水分子のラマン散乱断面積が、他の色素やフェニルチオール、DNA塩基などと比べて、ずっと小さい(~1/30)ことによると考えられる。我々は、flocculation-SERS法において、AgNP間ナノギャップでのSERS増強度は10⁶ -10¹⁰ 倍と、表面粗さのLSPよりずっと大きいことを利用して、水和金属イオンの捕捉と検出を試みた。その結果、ハロゲン化物で置換して負電荷を持たせたAgNP表面と水和金属イオンの静電的相互作用を利用して、AgNP間のナノギャップに水和金属イオンを捕捉し、溶媒和している水分子を初めて検出することに成功した[35]。このとき、濃厚塩水溶液の水分子の3200-3400 cm⁻¹⁰ のラマンスペクトルとは異なり、3500-3600 cm⁻¹⁰ の高波数に、水素結合が弱められた水分子が観測された。興味深いことに、このラマンバンドのピーク波数は、金属イオンの種類とともに、ハロゲン化物イオンの種類に依存して有意の違いを与えた。この結果から、AgNP間ナノギャップで水和金属イオンは、AgNP-X-..H2O..M+..H2O..X-AgNPの構造のsolvent shared ion pair(SIP)を形成していることが明らかになった(図➋-22)。

同時に低波数領域に束縛回転振動及びM..OH2伸縮振動バンドが観測された。以上のように、AgNPを用いるflocculation-SERS法により、AgNP表面のハロゲン化物を用いて、溶液中の水和金属イオンを捕捉し、SIPイオン対を形成することを見出した。より詳細な金属イオンの水和構造の解析を進めている。
4-4. 各種チオールの吸着状態の溶液組成依存性
4-5.生体分子の吸着状態分析
次の例は、flocculation-SERSの生体分子への適用結果である。複雑系である生体高分子の分光測定の意味・意義は、単なる還元主義ではなく、そもそも現時点では、複合した生体高分子の複雑な高次構造と機能の関係を追う(ことしかできない)段階であるというだけで、将来的には活性点の分子レベルの反応解析だけでなく、全分子状態の解析が可能になるはずである。その意味で、トップダウン的にマクロからミクロ、更にナノ・サブナノレベルへと分析レベルのダウンサイジング(X線・電子線回折、電子散乱による三次元構造塩基など特定の化学種の位置決定塩基配列・アミノ酸配列周囲の化学種との相互作用解析)が必要である。同時に、ボトムアップ方向に、生体高分子の構成要素である、DNA塩基、RNA塩基、アミノ酸から、ヌクレオチド、ペプチド、DNA, タンパクへと個々の分子の存在状態(構造、配向性、隣接分子との相互作用)の解析法を高次構造へと進めていく必要がある[36]。その意味で、SERS・SEIRA分光で、我々はボトムのDNA塩基、RNA塩基のflocculation-SERS測定を行っている。これまでに、AuNP表面への塩基分子の吸着状態のpH依存性、塩濃度依存性を測定することで、5つの塩基分子の吸着性が、1級アミノ基のあるなし及びプリン環を持つ(Adenine (A), Guanine (G))かピリミジン環を持つ(Cytosine (C), Thymine (T), Uracil (U))かにより系統的に区別できることを見出した[37, 38]。また、AuNPは通常クエン酸還元法で形成される(cit-AuNP)が、AuNP
表面に必ず残留するクエン酸が、一方ではその負電荷の為に分散液中でAuNPを孤立安定化し、もう一方では、DNA塩基のような吸着性の弱い化学種の吸着に大きな影響を与えることを確かめた。クエン酸を別の化学種に置き換えるのは、チオール以外では非常に困難であり、AuNPを細胞内に導入する場合も、細胞内での凝集や、マーカー分子の修飾の妨害となることなどの点で、注意しなければならない。他方で、クエン酸還元法によらない、例えばソリューションプラズマ法で形成したAuNP(SP-AuNP)は、as-prepared状態では表面がAuOx酸化物になっており、pHに敏感な表面電荷をもつ。さらに、SP-AuNPはcit-AuNPと同様のDNA塩基の吸着性を持つ一方で、cit-AuNPとは異なり、ほぼ完全にハロゲン化物置換できる。現在DNA塩基に対する吸着特性の詳細を測定中であるが、今後、SP-AuNPの表面修飾体が、より幅広く生体分子系に使われる可能性がある。さらに我々は、AuNPを用いたflocculation-SERS法の細胞内への適用を進めている。まず、細胞内にAuNPがエンドサイトーシス等で取り込まれる過程における、脂質膜、膜タンパク、膜のホスファイチジルコリンのリン酸基などとneat AuNP及び表面修飾したAuNPの相互作用解析から始めている。
参考文献
(25) Max Born, Emil Wolf (著), 草川 徹 (訳), 「光学の原理 第7版 Ⅰ- Ⅲ」, 東海大学出版会, 2005.
(26) P. C. Lee and D. Meisel, J. Phys. Chem. 1982, 86, 3391-3395.
(27) a) Frens, G. Nat. Phys. Sci. 1973, 20, 241. b) J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot, and A. Plech*, J. Phys. Chem. B 2006, 110, 15700-15707および引用文献.
(28) J.N. イスラエルアチヴィリ, 「分子間力と表面力 第3版」朝倉書店 2013.
(29) a) S. Funkner, G. Niehues, D. A. Schmidt, M. Hyeden, G. Schwaab, K. M. Callahan, D. J. Tobias, M. Havenith, J. Am. Chem. Soc. 2012, 134, 1030-1035, b) J. Gujt, M. Bešter-Rogač, B. Hribar-Lee, J. Mol. Liq. 2014, 190, 34-41.
(30) Y. Chen, J. L. Fulton, W. Partenheimer, J. Solution Chem. 2005, 34, 993-1007.
(31) S. Gopalakrischnan, D. Liu, H. C. Allen, M. Kuo, M. J. Shultz, Chem. Rev. 2006, 106, 1155-1175.
(32) C. J. Fennell, A. Bizjak, V. Vlachy, K. A. Dill, J. Phys. Chem. B 2009, 113, 6782-6791.
(33) M. Smiechowski, J. Stangret, Pure Appl. Chem. 2010, 82, 1869-1887.
(34) J-F. Li, Y-F. Huang, S. Duan, R. Pang, D-Y. Wu, Bin Ren, X. Xu and Z-Q. Tian, Phys. Chem. Chem. Phys., 2010, 12, 2493–2502.
(35) R. Kuwana, S. Handa, M. Futamata, Chemical Physics Letters in press 2018. (36) K. Watanabe, A. F. Palonpon, N. I. Smith, L. Chiu, A. Kasai, H. Hashimoto, S. Kawata, K. Fujita, Nature Comm. 2015, 6, 10095.
(37) T. Mukaiyama, T. Yajima, and M. Futamata, e-J. Surf. Sci. Nanotech. 2015, 13, 223-230.
(38) M. Seki, T. Mukaiyama, M. Futamata, submitted.